

Ovarian
cancer is the most lethal gynecological malignancy., the seventh most common cancer in women and the 18th most common cancer overall, with over 200,000 new cases worldwide each year.
Using epigenomic analysis
of patient tumors we have found common DNA methylation patterns in the most common subtype of ovarian cancer, high-grade serous ovarian cancer (HGSOC)., which has the highest mortality rate. The mortality rate is high because patients are generally diagnosed in an already advanced stage
Epigenetic modifications like DNA methylation are known to frequently affect gene regulation
involved in cancer-related processes but since epigenetic alterations are
reversible in nature, these changes have emerged as attractive targets for
epigenetic therapy for cancer. Yet modeling is a great challence in preclinical research. [1]
Recent genomic analyses showed that most commonly used HGSOC cell lines, like SKOV3 and A2780, are less representative models of HGSOC. Recently, patient-derived tumor xenografts (PDXs) i.e., patient tumor tissues transplanted directly into immune-deficient mice have appeared as better representative preclinical models than cell lines-based xenograft models. They recapitulate histological type, maintain genomic features and the reminiscent heterogeneity of the corresponding patient's primary tumor. [2]. Furthermore, treatment results of ovarian cancer PDXs have a good predictive value for standard platinum-based chemotherapy and novel therapeutic agents.
Recently, a panel of about 50 ovarian cancer PDXs with different histological subtypes have been established and genomically characterized for future cancer research at University Medical Centre Groningen, The Netherlands [3]. Although several comparative gene expression and mutational studies have been performed for HGSOC PDXs, comparable studies on the epigenome are not yet available. 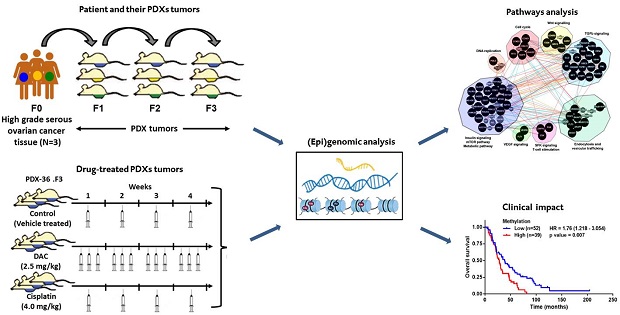
Credit:
doi 10.1186/s13073-016-0361-5
In a recent published report, my colleagues and I performed a genome-wide analysis of the DNA methylome of HGSOC patients with their corresponding PDX tumors, from different generations of tumor propagation [from patient tumor (F0) to first mouse (F1) and from that mouse (F1) to another mouse (F2) and so on (F3) as shown in the figure]. We found minor changes of 0.6-1.0% of all analyzed genome-wide methylation-enriched regions (CpG sites) during propagation showing that HGSOC PDXs were epigenetically stable [4]. Further, they analyzed global methylome changes after treatment of HGSOC PDXs with clinically used DNA demethylating agent decitabine (DAC) and cisplatin, as platinum-containing chemotherapy which is regarded as standard care in first-line treatment of HGSOC.
Treatment of PDXs with decitabine caused a reduction in methylation in 10.6% of CpG sites in comparison to untreated PDXs. Unlike to DAC treatment, cisplatin treatment had a marginal effect on the PDX methylome. To gain more insight of DAC-induced DNA methylation changes, we performed biological pathway analysis of decitabine-treated PDX tumors. This analysis revealed several putative epigenetically regulated pathways (e.g. Src family kinase pathway). Particularly, C-terminal Src kinase (CSK) gene was successfully validated for its epigenetic regulation in different PDX models and various ovarian cancer cell lines. Using publicly available patient (epi)genomic datasets, we showed that low CSK methylation and high CSK expression are both highly associated with improved progression-free survival and overall survival in HGSOC patients.
University Medical Center Groningen
HGSOC PDXs resemble the global epigenome of patients over many generations of propagation and can be modulated by epigenetic drugs and epigenetically regulated genes such as CSK and related pathways were identified in HGSOC. Our results match epigenomic studies on PDX models of various other cancer types like colon, lung, head&neck and osetosarcome, so it's an exciting time PDXs for cancer epigenome studies.
Citation: Tomar T, de Jong S, Alkema NG, Hoekman RL, Meersma GJ, Klip HG, et al. Genome-wide methylation profiling of ovarian cancer patient-derived xenografts treated with the demethylating agent decitabine identifies novel epigenetically regulated genes and pathways. Genome Med. Genome Medicine; 2016;8: 107. doi:10.1186/s13073-016-0361-5
References:
1. Alkema NG, Wisman GBA, van der Zee AGJ, van Vugt MATM, de Jong S. Studying platinum sensitivity and resistance in high-grade serous ovarian cancer: different models for different questions. Drug Resist Updat. Elsevier Ltd; 2016;24: 55–69. doi:10.1016/j.drup.2015.11.005
2. Hidalgo M, Amant F, Biankin A V, Budinska E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4: 998–1013. doi:10.1158/2159-8290.CD-14-0001
3. Alkema NG, Tomar T, Duiker EW, Jan Meersma G, Klip H, van der Zee AGJ, et al. Biobanking of patient and patient-derived xenograft ovarian tumour tissue: efficient preservation with low and high fetal calf serum based methods. Sci Rep. Nature Publishing Group; 2015;5: 1–12. doi:10.1038/srep14495
4. Tomar T, de Jong S, Alkema NG, Hoekman RL, Meersma GJ, Klip HG, et al. Genome-wide methylation profiling of ovarian cancer patient-derived xenografts treated with the demethylating agent decitabine identifies novel epigenetically regulated genes and pathways. Genome Med. Genome Medicine; 2016;8: 107. doi:10.1186/s13073-016-0361-5

Epigenetic Predictors Of Ovarian Cancer
Related articles
- Results In Phase I Trial Of OMP-54F28, A Wnt Inhibitor Targeting Cancer Stem Cells
- No BRCA1 Gene Needed: New Test Predicts The Risk Of Non-Hereditary Breast Cancer
- Epigenetic Signatures Consistent In Metastases Of 13 Prostate Cancer Victims
- Epigenetics: New Tool For Precision Medicine
- Genome-Wide DNA Methylation And Transcriptomes At Single Base Resolution

Comments